Dynamique de la chromatine et expression des gènes
Un objectif majeur de l’équipe CRP est de caractériser la relation entre la configuration du chromosome et l’expression du génome à l’échelle globale, en se basant sur le modèle de bactérie phytopathogène Dickeya dadantii. Ce mode de régulation "analogique", actuellement sous-estimé et presque absent des travaux de modélisation, diffère de la régulation classique "digitale", d'une part par les acteurs impliqués (NAPs et surenroulement de l'ADN vs facteurs de transcription) et d'autre part par son action globale sur le génome. Les méthodes utilisées sont la transcriptomique, la manipulation du chromosome, et la modélisation computationnelle et biophysique.
1. Caractérisation des contributions analogue et digitale en régulation transcriptionnelle
Pour comprendre le réseau de régulation responsable de l'expression des gènes de virulence et donc du succès de l'infection, nous utilisons de nouvelles approches d'analyse globale de l'expression des gènes, qui établissent des corrélations entre les propriétés physico-dynamiques des séquences des gènes exprimés (énergie de melting, sensibilité à la relaxation...) et les effets de structuration de l'ADN par les Protéines Associées au Nucléoïde (NAPs). Cette approche systémique a permis de démontrer, chez Dickeya dadantii, l'existence de domaines chromosomiques où de nombreux gènes sont activés ou réprimés de façon cohérente en réponse à des stress environnementaux variés, probablement liés au repliement 3D du chromosome, tandis que les différences de réponses sont liées aux propriétés physico-chimiques des séquences d'ADN qui les encodent. La comparaison de nos données avec des profils d'expression in planta révèle l'impact combiné des facteurs de stress, et nous permet de prédire quel stress est rencontré par Dickeya dadantii durant les différentes phases de l'infection (Jiang et al. 2015 and 2016). En collaboration avec M. Hütt à l'université Jacobs (Bremen, Allemagne) et G. Muskhelishvili (Tbilissi, Géorgie), nous développons maintenant une analyse de données de transcriptomique, afin de distinguer les contributions analogique et digitale à la régulation de l'expression génomique, dans des conditions pertinentes pour l'infection et avec des souches mutantes de NAPs (Meyer et al. 2018, Muskhelishvili et al. 2019).
2. Dynamique du chromosome et activité transcriptionnelle à grande échelle
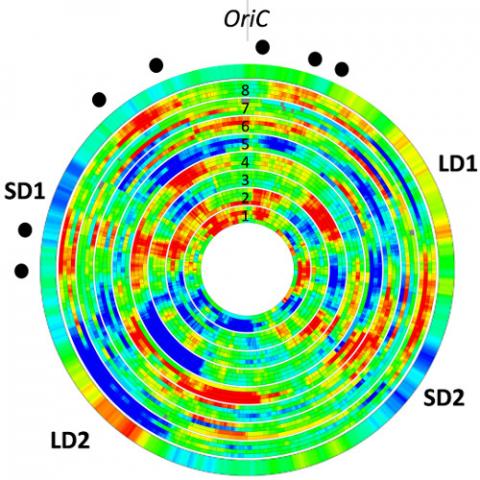
Des analyses précédentes ont démontré l'existence chez Dickeya de domaines chromosomiques d'expression cohérente, de grande taille et fluctuants (Jiang et al 2015). Nous testons maintenant l'hypothèse qu'à cette échelle, la dynamique du chromosome est couplée à son activité transcriptionnelle (sans doute dans les deux sens), ce qui pourrait être observé physiquement. Pour cela, nous collaborons avec l'équipe de Christian Lesterlin (IBCP, Lyon) en imagerie super-résolution de tags fluorescents dans des cellules vivantes. Le projet consiste à suivre la localisation de différents loci sur le chromosome, ainsi que la position des NAPs, dans différentes conditions où les domaines sont activés ou réprimés, et en utilisant différentes souches mutantes (avec des gènes de NAPs déplacés). L'hypothèse de travail est que l'organisation spatiale du chromosome es tliée à son état fonctionnelle, les régions exprimées étant typiquement déplacées vers la périphérie de la cellule, et l'objectif est de déchiffrer le rôle spécifique des NAPs dans ce mécanisme.
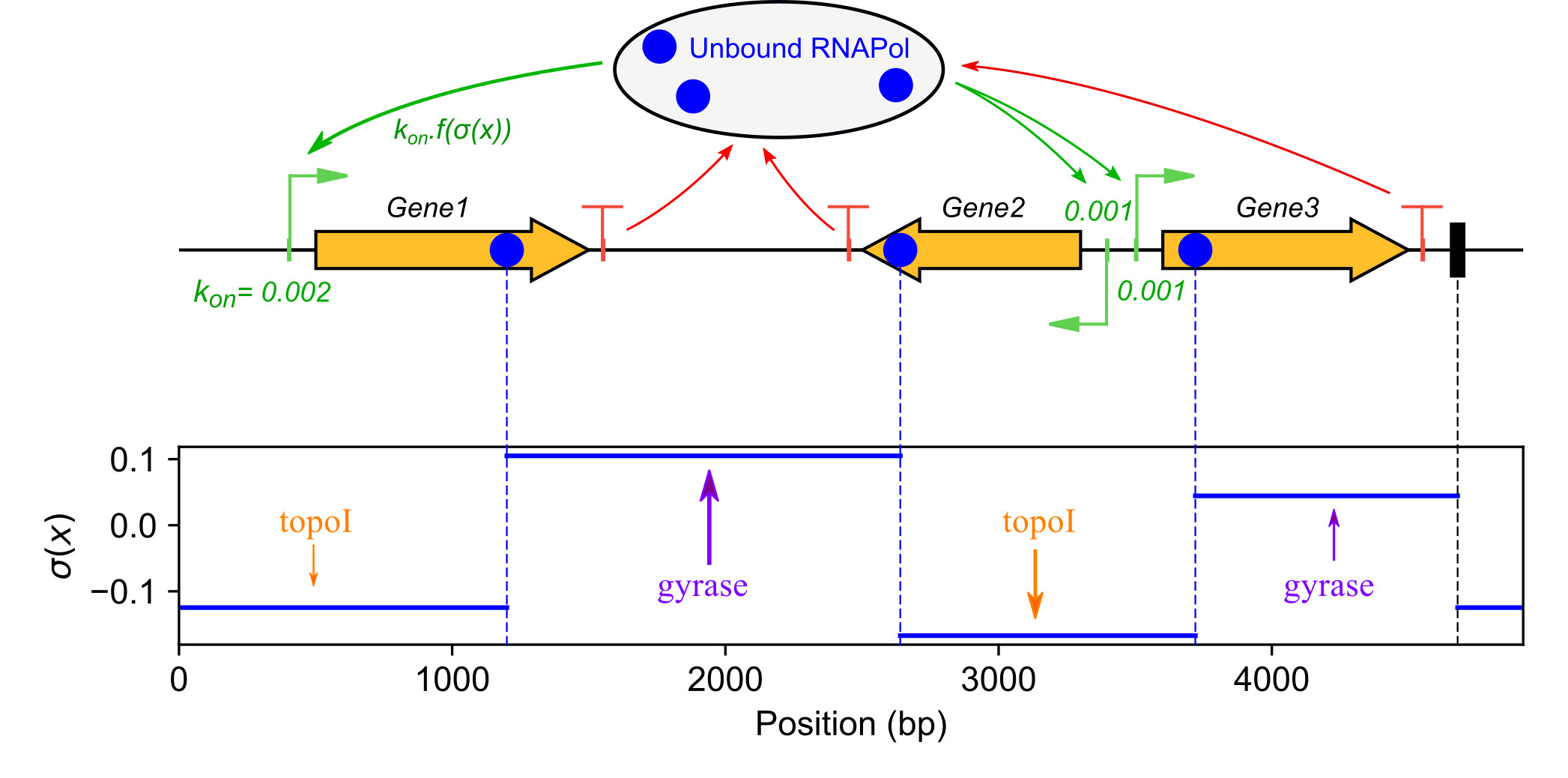
3. Régulation transcriptionnelle globale par le surenroulement de l'ADN
Nous développons des modèles quantitatifs de la régulation globale de la transcription bactérienne par le surenroulement de l'ADN (Martis et al. 2019). L'effet de ce dernier sur l'activité des gènes est expliqué en se basant sur (1) la structure de leur promoteur et (2) les variations spatiales du surenroulement le long du chromosome. L'initiation de la transcription est décrite à l'aide de modèles thermodynamiques de l'interaction ADN-ARN Polymérase, démontrant le rôle (i) de la taille du spacer entre sites -10 et -35, affectant la formation du complexe fermé, (ii) de la séquence du discriminateur (entre la boîte -10 et le TSS) affectant la dénaturation nécessaire à la formation d'un complexe ouvert. Ces hypothèses sont testées en utilisant des données de transcriptomique publiées et originales d'espèces phylogénétiquement distantes, et par des expériences plus spécifiques sur Dickeya. Les variations spatiales de surenroulement de l'ADN le long du chromosome de Dickeya sont analysées à l'aide de nouvelles données obtenues avec l'agent intercalant tri-méthyl-psoralen, en collaboration avec Monika Glinkowska (Univ. Gdansk, Pologne). Cette analyse est complémentée par un travail de modélisation (El Houdaigui et al. 2019), et ce projet bénéficie d'un financement ANR JCJC (LoToReg 2018).